化粧品製造販売業、化粧品製造業や医療機器製造販売業、医療機器製造業では
多数の手順書や品質標準書(製品標準書)に従って日々の業務が運営されています。
例)新たに手順書が必要になって制定しようとした時や手順書内容の修正をしようと改訂しようとした時
例)新しい製品の品質標準書(製品標準書)を制定しようとした時、修正をしようと改訂しようとした時
これらの文書の制定や改訂に関して間違った運用がされたり問題がある文書管理運用が見受けられます。
本来は文書管理手順書等で詳細に運用を決めてあるはずなのですが行政からアップされている化粧品製造販売業のモデル手順書がざっくりしか書いてないので間違いや理解出来ていない方がおられます。
●使用しなくなった文書や記録は保存年数が決まっています(最低でも何年以上と)。
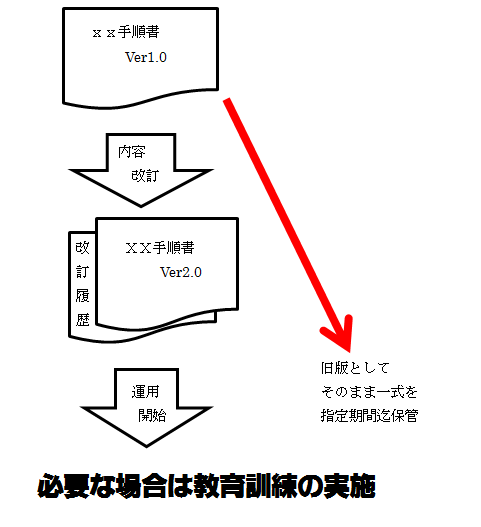
●手順書や標準書は版数管理をします(第1版とかVer1.0とか)。
●使用しなくなった旧版は同じ場所(最新版の後ろとか)に置いて(ファイリング)はダメです、違う所に保存してください。
●使用しなくなったの意味は文書や記録によって変わります。
1.たまに見かける間違った文書管理運用
版数管理を行う必要がある文書なのに初版の中身を変更している
↑
中身を変更改訂する場合は版数のアップを行い、前の版は使用しなくなった旧文書として保存する必要があります。
例)化粧品GQP手順書(Ver1)==>変更改訂を行ったものは化粧品GQP手順書(Ver2)となり運用される
化粧品GQP手順書(Ver1)は使用されなくなった旧文書として決められた保存年数の間は保存される(破棄してはいけない)
もちろん、制定改訂履歴もつけないといけません。
(改訂した箇所や改訂した理由を書きます)
手順書等の文書の作成者、確認者は記入されているが承認者がない
↑
会社としての手順書等ですから最終的に会社として承認されていないと正式発行された事になりません、代表者(代表取締役)とか責任役員とかの承認が必要でしょう。
2.問題があると思われる文書管理運用
関連部署の確認、承認がされていない
●制定(改訂)する前に、関連部署に確認をとる必要があるでしょう。
●完成したら、関連部署から承認を得る必要があるでしょう。
教育訓練されていない
手順書、標準書が制定、改訂されたら、その内容を関連部署に周知するために教育訓練が必要になる場合があります。
この教育訓練をしていないと、昔のやり方とかで作業していた等の問題が発生したりしますので注意です。
教育訓練なんて書くと大げさですが、周知徹底されれば良いんです。
ようするに、全て手順書などには関連部署があるでしょってことです。
手順書、標準書やその記録様式を作成したり変更したりする場合に作成者(又は変更者)が勝手にやってはいけないって事です、それに関連する部署(営業だったり製造だったり業務だったり)に許可、確認を貰い、周知する事です。
たまに、文書の改訂履歴だけがあり、作成者と承認者の印だけってのがあります。
(たいてい、そのような会社では作成者と承認者だけで改訂してしまって、実作業と違っている場合が多いです)
これでは関連部署へ確認がされたこと、周知されたことを記録されていませんよね、
手順書、標準書と実際の運用の相違が生まれる一番の原因がこれです。
管理工程図のようなものを作成して、業務フローを明確にしておくのも手だと思います。
不明点等ありましたら、お問い合わせから 質問して下さい。
