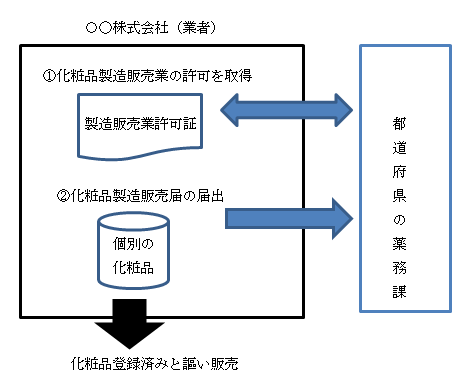
化粧品製造販売業、製造業
医療機器製造販売業、製造業
これらの業務を色々な手順書や品質マニュアルに従って行うのですが、
運用で一番重要なことは何か?って考えてみました。
まず、
「うちは自由な社風でルールを作らないので、手順書なんて必要ない~」とか
「うちはフラットな組織なので権限を現場に委譲してます~」
なんてのは、薬機法(旧薬事法)がからむ業界には向いていないと思います。
(残念ですが、IT業界などそれが許される業界にいて下さい、新規事業としてこの業界に入る事を考えない方が今現在の組織のためです)
今時の方には嫌われるかもですが、
トップダウンの階層組織で全ては手順書に従い行動し、常に上長の確認、承認なしには何も行動出来なくて、もしそれが行われていない事が見つかれば、監査において重大欠陥として指摘されますし、最悪は製品回収などに発展する事も考えられます。
そうです、上のハンコ、上のハンコ、上のハンコ・・・と言う嫌われる組織なのです(ISOでも同じ組織ですが)
本題に入りますが、手順書等の運用で一番重要な事はなにか?
全ての基本は、文書の管理と記録の管理にあると思います。
例えば、医療機器製造販売業の許可申請を行った時に
現地調査(実地調査)に先立ち手順書等を薬務課に送るのですが、
その時の薬務課から要求されるのは、品質マニュアルと文書管理の手順書だったりします。
また、PMDAから出ている
QMS適合性調査における指摘事例及び適合に向けての考え方について(2017年版 )
https://www.pmda.go.jp/files/000217332.pdf
においても、文書の管理、記録の管理が指摘事項として多いことが記述されています。
1.全ての行動は、手順書と呼ばれる文書に従って行い
2.行った行動の結果を記録に残す。
単純に言えばこれだけです。
その行動(行わなければならない行動)とは、薬機法(旧薬事法)、QMS省令で指定され縛られたものになります。(経理業務を薬機法で縛ったりしませんからね)
その行動を縛る手順書は誰が作成し、誰が確認し、誰が承認したのか。
また、その行動を行う人々に最新の手順を周知徹底しなければなりません。
行動の内容を変えたい場合には、その行動を縛る手順書を先に変えてから行動を起こすようにしなければなりません。
ここで注意したいのは、手順書やその記録様式を作成したり変更したりする場合に作成者(又は変更者)が勝手にやってはいけないって事です、それに関連する部署(営業だったり製造だったり)に許可、確認を貰う事です。
たまに、文書の改訂履歴だけがあり、作成者と承認者の印だけってのがあります。これでは関連部署へ周知確認がされていません。内部情報伝達ってところが抜けている事になります。
上記から、
まずは、文書管理(記録管理)手順書から勉強する必要があると思います。
見た目だけで全てをPC入力にしていませんか?
うちは入力ミスしませんから、手書きなんてカッコ悪い なんて理由で1つの様式に全てをword等で入力して記録を作成しているところがあります。
他社で手書きの記録を見た事が無い なんて言う方が居ましたが、その他社さんは電子認証システム等を導入しているんです、印刷物だけで判断してはいけません。
電磁的記録が認められるのは、真正性、見読性、保存性等適切な条件が確保できる場合です。
*たまにですが、A=>B=>C と行動が流れていく記録なのに、全てwordで行動内容まで印刷された記録を見せてくる会社が有りますが、ワークフローシステムみたいな電子認証システムを導入してるんですか?
Aで作成した文書ではwordで印刷されていても、BやCではその印刷された記録の紙に手書きでしか書けないのですから。
もちろん、そのような電子認証システムを導入していれば問題ないのです。
この辺りはIT系の人材でないと理解できないので難しいところでは有るのですが
IT系の方でも地方大学の情報系の大学教員など、文書と記録の違いが分からなくて記録が手書きでないとダメと大学から言われた~と反発するなんて恥ずかしいツイートしゃう位なので難しい話です。 (ISOの監査員でもIT系の審査したことの無い審査員だと理解できていない審査員もいました)
あるところからのコピペですが求められる要件を書いておきます、wordなどで簡単に実現出来るものでは無い事を理解していただければと思います。
真正性とは??
真正な記録とは、次のことを立証できるものである。
記録が主張しているとおりのものであること(本物)
それを作成又は送付したと主張するものが、作成又は送付していること
主張された時間に作成し、送付していること
記録が真正であることを確実にするために、組織は、記録作成者に権限を与え、それが誰かを明確にすること。
許可の無い記録の追加、削除、変更、利用及び隠蔽から確実に記録が守られるように、記録の作成、取得、送信、維持及び処分を管理する方針及び手順を実施し、文書化すること。
ER/ES指針において、真正性が要件になっている理由は、電磁的記録および電子署名には下記のようなリスクがあり、それらリスクを回避しなければならないからである。
電磁的記録の作成者がわからなくなるリスク
電磁的記録の承認者がわからなくなるリスク
許可されていない者が電磁的記録の入力・変更を行うリスク
電磁的記録を上書きされてしまうリスク
電磁的記録を削除されてしまうリスク
電磁的記録および電子署名を改ざんされるリスク
電磁的記録および電子署名を誤って変更・削除してしまうリスク
不適切な者へ権限を与えるリスク
パブリックコメントの回答(No.58)によると、真正性はAuthenticityの訳とある。
しかしここでは「電磁的記録が完全、正確であり、かつ信頼できるとともに、作成、変更、削除の責任の所在が明確であること。」 と記載されている。この文章を読む限り、真正性は、米国版Part11のIntegrityとAuthenticityを兼ね備えたものであるといえる。 なぜならばIntegrityとは「完全、正確であり、かつ信頼できる」ということであり、Authenticityとは「責任の所在が明確であること」だからである。
1999年5月に公表された「Compliance Policy Guide 7153.17」によると、FDAでは、 Integrityを重要視してPart11査察を行うとある。 Part11施行後のWarning LetterもやはりこのIntegrityに関するものが過半数を占めている。
完全とは、その内容が完結していて変更されていないことを意味する。 記録は許可の無い変更から守られなければならない。また生データのみではなく、タイム・スタンプや変更履歴等のメタデータが正確にリンクされている状態を指す。 (FDAでは、このことに関して、データの品質要件として、 Attributableという用語を使用している。)
信頼できるとは、システムがセキュリティなどにより、改ざんから守られており、さらに変更などの記録がとられている状態を指す。
不明点等ありましたら、お問い合わせから 質問して下さい。